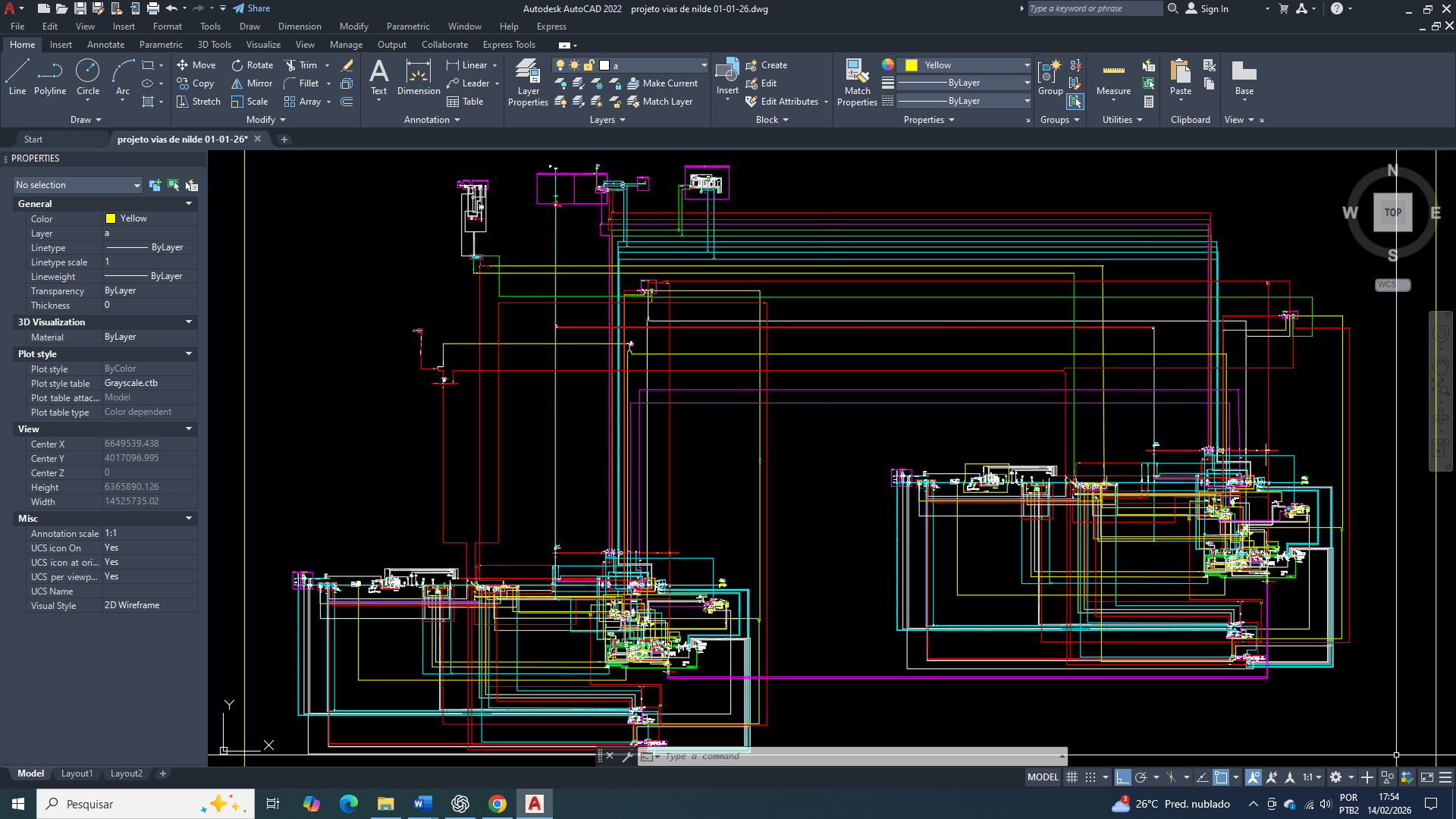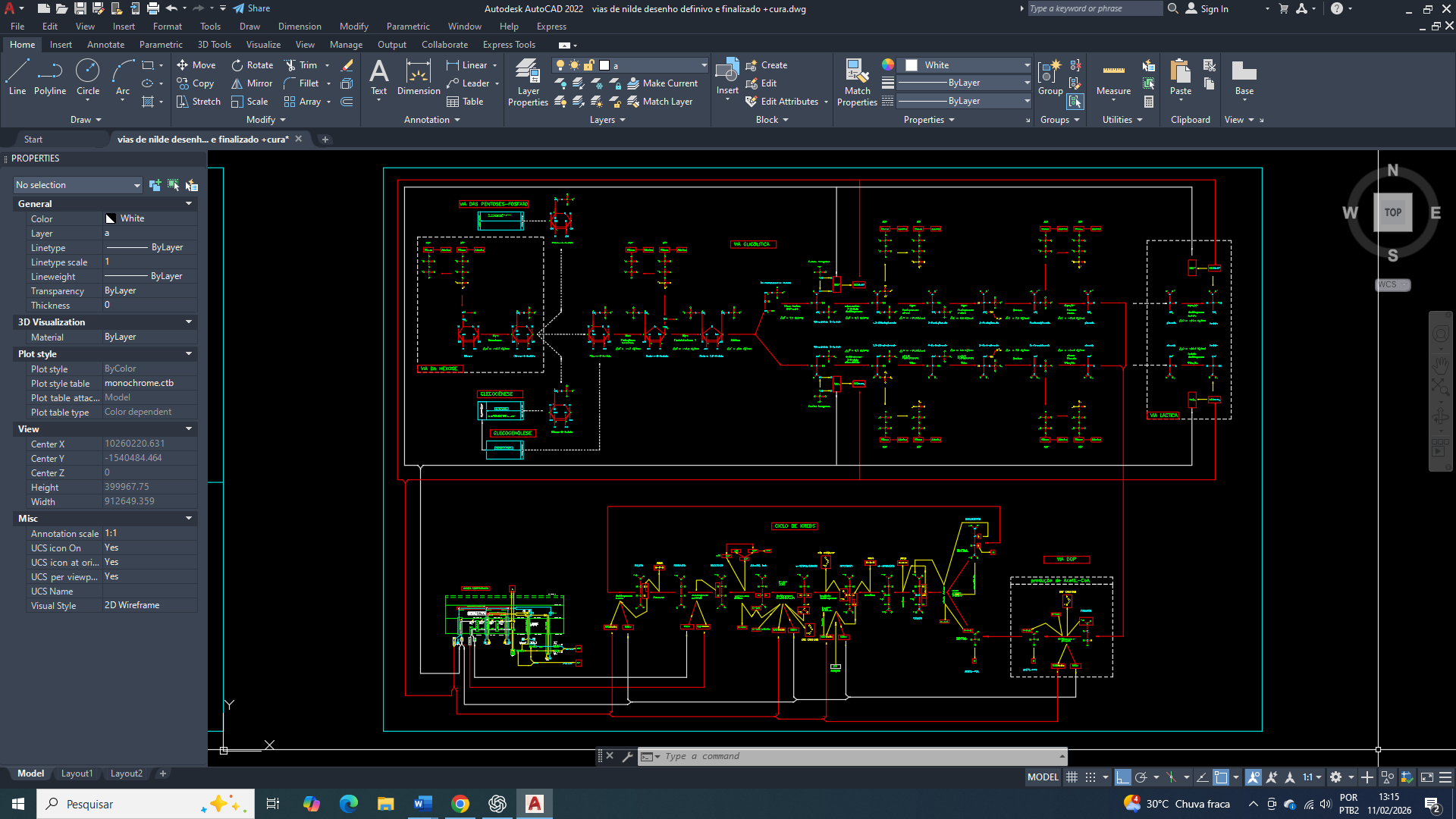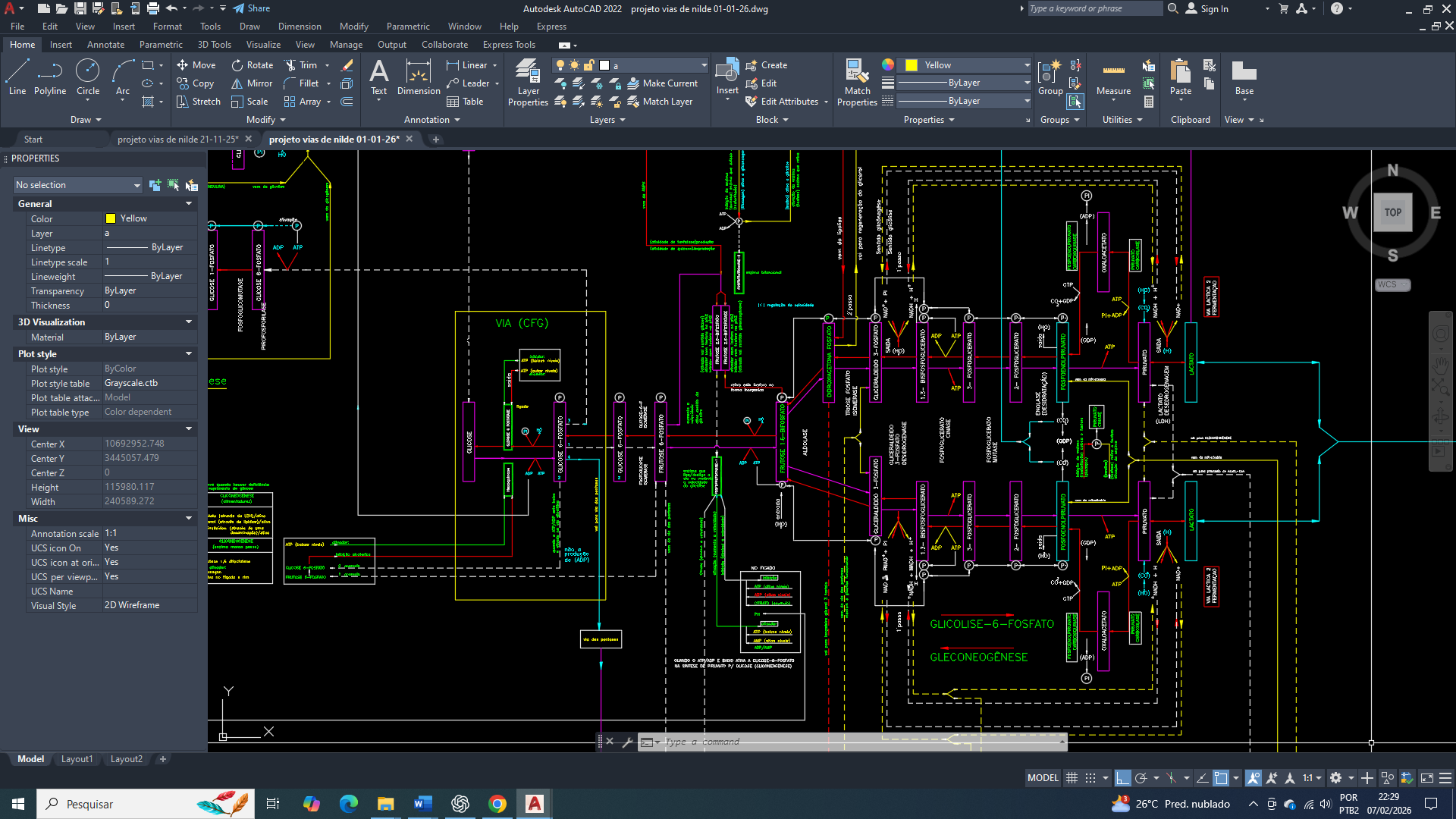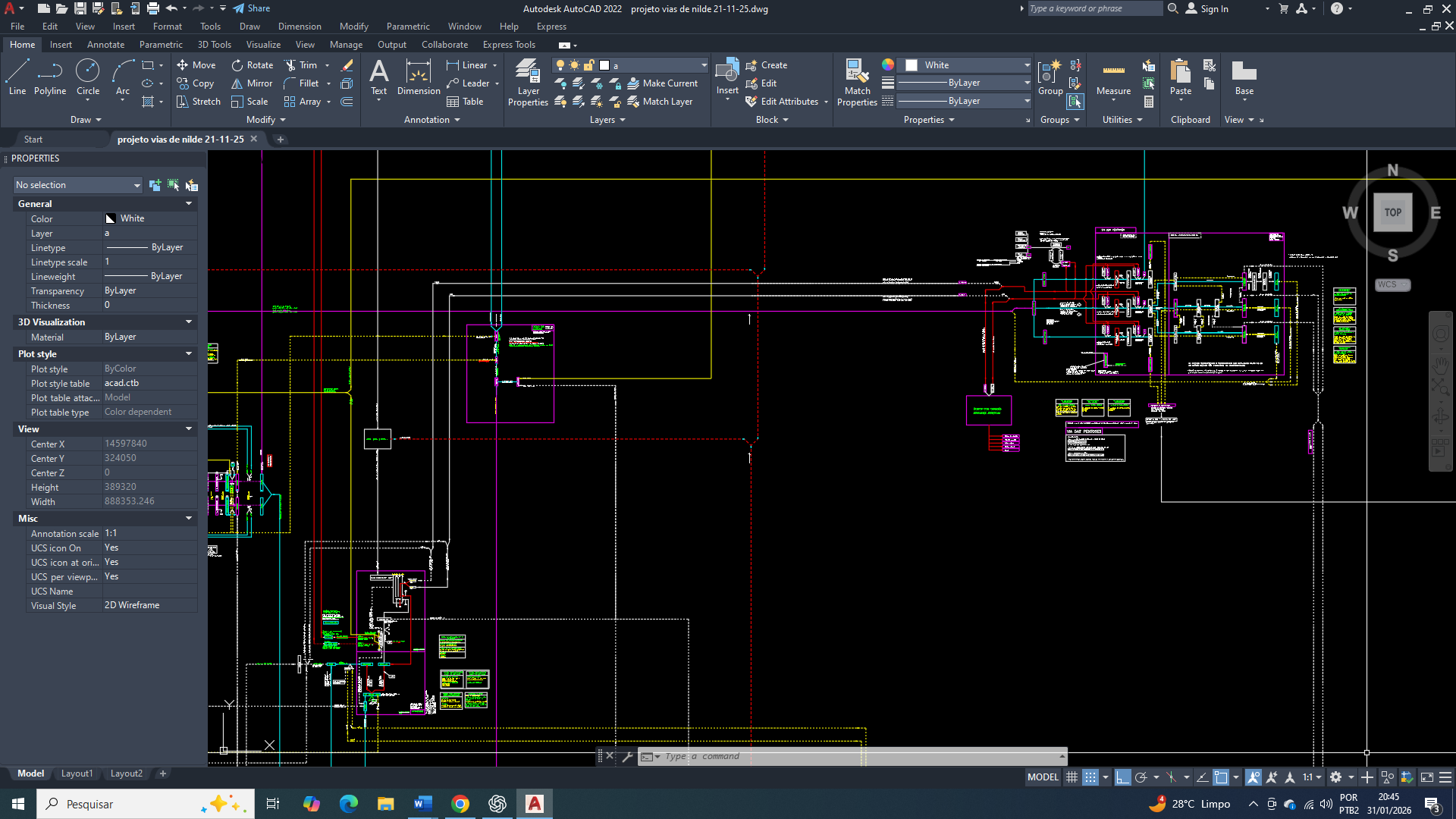
The cure for breast cancer: 16 – biochemical control of acetyl-CoA carboxylase in the context of metabolic reprogramming, AMPK, and insulin signaling
Parte 16 da série: Reprogramação Metabólica no Câncer de Mama
Fabio Henrique Amaral de Almeida
Pesquisador independente (Biomedicina), São luís, MA- Brasil
Endereço para correspondência (Para ajudar financeiramente a manter este canal)
Pix. 303 278 223 68
E-mail: ftorpedo3@gmail.com
postado em: 20/01/2026
revisado em:
Agradecimento.
Exclusivamente a DEUS.
Que me permite, por sua vontade, a sabedoria e o entendimento da verdade, assim como a todos aqueles que Ele julgar terem esse direito.
Resumo
No câncer de mama, a reprogramação do metabolismo lipídico constitui um dos pilares para a sustentação da proliferação celular, manutenção da integridade de membranas e geração de sinais pró-tumorais. A acetil-CoA carboxilase (ACC), enzima limitante da lipogênese de novo, ocupa posição estratégica nesse processo ao converter acetil-CoA em malonil-CoA. Sua atividade é rigidamente controlada por modificações covalentes e por mecanismos alostéricos, destacando-se a fosforilação inibitória mediada pela AMPK e a desfosforilação promovida por fosfatases ativadas pela insulina, principalmente a PP2A. Embora a AMPK represente um potente freio energético à lipogênese, células de câncer de mama frequentemente mantêm a ACC funcionalmente completamente ativa por meio da sinalização insulínica/IGF-1, reorganização subcelular de fosfatases e abundância citosólica de acetil-CoA. Este trabalho descreve, de forma integrada e bioquimicamente fundamentada, os mecanismos que governam a atividade da ACC no câncer de mama, destacando a hierarquia regulatória entre estado energético, sinalização hormonal e compartimentalização metabólica.
Palavras-chave: câncer de mama; ACC; AMPK; insulina; PP2A; lipogênese; reprogramação metabólica.
Abstract
In breast cancer, lipid metabolic reprogramming represents a central axis sustaining cellular proliferation, membrane biosynthesis, and oncogenic signaling. Acetyl-CoA carboxylase (ACC), the rate-limiting enzyme of de novo lipogenesis, plays a pivotal role by catalyzing the conversion of acetyl-CoA into malonyl-CoA. ACC activity is tightly regulated by covalent modifications and allosteric mechanisms, notably inhibitory phosphorylation by AMP-activated protein kinase (AMPK) and dephosphorylation mediated by insulin-activated phosphatases, primarily protein phosphatase 2A (PP2A). Although AMPK functions as a major energetic brake on lipogenesis, breast cancer cells frequently preserve ACC activity through insulin/IGF-1 signaling, subcellular redistribution of phosphatases, and sustained cytosolic acetyl-CoA availability. This article provides an integrated and biochemically grounded analysis of the regulatory mechanisms governing ACC function in breast cancer, emphasizing the hierarchical interplay between energetic status, hormonal signaling, and metabolic compartmentalization.
Keywords: breast cancer; ACC; AMPK; insulin; PP2A; lipogenesis; metabolic reprogramming.
Introdução
A reprogramação metabólica é uma característica universal das células cancerígenas e vai muito além do aumento da glicólise aeróbia. No câncer de mama, observa-se uma profunda reorganização do metabolismo do carbono, com um direcionamento preferencial de intermediários glicolíticos e mitocondriais para a biossíntese lipídica. Onde esse redirecionamento atende à elevada demanda por fosfolipídios, ácidos graxos saturados e sinais lipídicos necessários à proliferação, sobrevivência e invasão tumoral.
E nesse contexto, que a acetil-CoA carboxilase (ACC) emerge como um dos pontos de convergência entre o estado energético celular, a sinalização hormonal e a disponibilidade de substrato. A atividade da ACC não é determinada apenas pela concentração de acetil-CoA, mas por um sistema hierárquico de controle que integra sensores energéticos (AMPK), fosfatases reguladas por insulina (PP2A) e organização espacial dos complexos lipogênicos. Compreender esse controle é essencial para explicar como células de câncer de mama mantêm a lipogênese ativada mesmo em ambientes de estresse metabólico.
Ativação permanete da lipogenese no câncer de mama
A acetil-CoA carboxilase catalisa a carboxilação do acetil-CoA em malonil-CoA, reação dependente de ATP e biotina que define a velocidade da lipogênese de novo. Em células de câncer de mama, a produção citosólica de acetil-CoA encontra-se persistentemente elevada, principalmente devido ao aumento da exportação mitocondrial de citrato para o citosol e à atividade atualmente expressa da ATP-citrato liase, acopladas à alta taxa glicolítica que são características dessas células. No entanto, a simples abundância de acetil-CoA não é suficiente para determinar a atividade da acetil-CoA carboxilase(ACC), uma vez que essa enzima está subordinada a um sistema hierárquico de controle no qual a modificação covalente imposta pela AMPK ocupa posição dominante. Em condições de estresse energético, refletidas por alterações nas razões AMP/ATP ou ADP/ATP, ou ainda por elevação da concentração de cálcio intracelular via CaMKKβ, a AMPK é ativada e fosforila a acetil-CoA carboxilase(ACC) em resíduos críticos, promovendo uma inibição conformacional que reduz sua capacidade catalítica, impede a polimerização funcional da enzima e diminui sua afinidade por cofatores essenciais. Essa fosforilação inibitória não pode ser revertida por ativadores alostéricos como o citrato nem por aumento da concentração de substrato, estabelecendo a primazia do estado energético sobre a biossíntese lipídica.
Em oposição direta a esse controle, a insulina atua como um potente sinal anabólico ao promover a desfosforilação da acetil-CoA carboxilase(ACC) por meio da ativação de fosfatases serina/treonina, principalmente a PP2A, após a ativação do eixo receptor de insulina–PI3K–Akt. A insulina não fosforila a acetil-CoA carboxilase(ACC), mas remove o sinal inibitório imposto pela AMPK, restaurando a conformação cataliticamente ativa da enzima e permitindo a retomada da produção de malonil-CoA. Um aspecto crucial desse processo é que a PP2A não atua de forma homogênea no citosol, mas é recrutada para microdomínios específicos por meio de subunidades regulatórias que determinam a sua localização subcelular. No câncer de mama, há evidências consistentes de que redistribuição da PP2A para regiões citosólicas enriquecidas em enzimas lipogênicas, criando populações de PP2A local que desfosforilam a acetil-CoA carboxilase(ACC) de maneira altamente eficiente, mesmo na presença de atividade global da AMPK.
Esse arranjo espacial permite que a acetil-CoA carboxilase(ACC) permaneça funcionalmente ativa em nichos lipogênicos específicos, sustentando a síntese de ácidos graxos necessários ao crescimento tumoral. A abundância citosólica de acetil-CoA, embora incapaz de superar diretamente a fosforilação inibitória da acetil-CoA carboxilase(ACC), garante que, uma vez desfosforilada, a enzima opere em um regime de fluxo elevado, conferindo grande plasticidade metabólica às células tumorais. Além disso, o acetil-CoA excedente vai alimenta processos paralelos, como a acetilação de histonas e a remodelação epigenética, reforçando programas transcricionais pró-tumorais. O resultado é um estado metabólico híbrido, no qual o freio energético que é imposto pela AMPK não é abolido, mas é funcionalmente contornado por sinais hormonais e por mecanismos de compartimentalização enzimática, isso permiti que a lipogênese de novo persista em níveis compatíveis com a progressão do câncer de mama.
A uma aparente contradição das funções da AMPK × insulina no câncer de mama.
O ponto central para compreender a coexistência funcional entre AMPK e insulina no câncer de mama é reconhecer que, nesse contexto, a ativação da AMPK deixa de ser predominantemente consequência de um déficit energético.
Em células normais, a AMPK é classicamente ativada por aumento da razão AMP/ATP ou ADP/ATP, refletindo queda na carga energética celular. No entanto, em células de câncer de mama operam majoritariamente sob um regime de glicólise-6-fosfato fermentativa acelerada, no qual a produção de ATP citosólico se torna elevada e contínua, mesmo na presença de oxigênio. Essa alta taxa glicolítica é suficiente para manter os pools globais de ATP em níveis que, isoladamente, não sustentariam a ativação da AMPK por estresse energético clássico. Esse é o ponto (x) da questão, pois dessa forma, o eixo AMP/ATP perde protagonismo como gatilho dominante da AMPK nesse cenário tumoral.
Apesar disso, a AMPK não se torna inativa. Sua ativação passa a ocorrer majoritariamente por vias alternativas, especialmente pela elevação crônica do cálcio intracelular, que é uma característica bem documentada do câncer de mama. Alterações na homeostase de Ca²⁺, decorrentes de remodelação de canais iônicos, sinalização por receptores acoplados à proteína G e estresse do retículo endoplasmático, tudo isso leva à ativação da CaMKKβ, que fosforila e ativa a AMPK independentemente do estado energético global. Sendo assim, a AMPK permanece funcional, mas agora integrada a um contexto fisiológico completamente distinto do seu funcionamento padrão para qual foi desenhada: pois nesse novo contexto ela não responde primariamente à escassez de ATP, mas a sinais de desorganização iônica e estresse celular.
Esse deslocamento funcional da AMPK é fundamental para entender por que sua ativação não resulta, necessariamente, na supressão efetiva da lipogênese no câncer de mama. A AMPK ativada por cálcio mantém a capacidade de fosforilar alvos metabólicos, incluindo a acetil-CoA carboxilase, impondo um freio potencial à lipogênese. Entretanto, em paralelo, a célula tumoral está imersa em um ambiente de forte sinalização anabólica, também mediada por insulina e IGF-1, com ativação sustentada pelo eixo PI3K–Akt–mTOR. Esse eixo não apenas promove a expressão de enzimas lipogênicas, como também ativa fosfatases serina/treonina, especialmente a PP2A, responsáveis por remover a fosforilação inibitória da acetil-CoA carboxilase(ACC).
A questão mais importante, portanto, não é se a AMPK está ativa ou não, mas onde e com que eficácia sua ação é mantida. A insulina prevalece sobre a AMPK em pontos específicos, como no caso da acetil-CoA carboxilase(ACC), porque atua no mesmo resíduo regulatório por um mecanismo espacialmente privilegiado. A proteína fosfatase 2 (PP2A) é recrutada para microdomínios lipogênicos citosólicos desfosforila a acetil-CoA carboxilase(ACC) de forma rápida e local, neutralizando o efeito da AMPK mesmo quando esta se encontra ativa em nível global. Dessa forma, a acetil-CoA carboxilase(ACC) passa a ter como efeito “padronizar ” mais a ação da fosfatase do que a da quinase, preservando sua atividade catalítica e sustentando a produção de malonil-CoA.
Esse arranjo explica por que, no câncer de mama, AMPK e insulina não se comportam como vias simplesmente antagônicas. A AMPK, ativada predominantemente por cálcio, continua a sinalizar estresse celular e a modular diversos processos adaptativos, enquanto que a insulina impõe prioridade anabólica em pontos que não estão agindo de maneira padrão, da programação metabólica, nesse caso a ativação da lipogênese. A prevalência da insulina sobre a ação clássica da AMPK, na acetil-CoA carboxilase(ACC) não decorre de uma falha da AMPK, mas de uma reorganização hierárquica e espacial do controle metabólico, na qual a compartimentalização de fosfatases e a abundância de acetil-CoA garantem que a lipogênese permaneça funcional mesmo em um ambiente de estresse sinalizado pela AMPK.
Em síntese, o câncer de mama não elimina a função da AMPK; ele a desloca a sua função energética clássica e, simultaneamente, cria mecanismos locais já existentes, que neutralizam seus efeitos sobre alvos críticos como a acetil-CoA carboxilase (ACC). Esse contexto híbrido é o que permite a coexistência de alta produção de ATP glicolítico, ativação de AMPK por cálcio e predominância funcional da insulina na sustentação da lipogênese tumoral.
Uma interpretação diferenciada no câncer de mama
No câncer de mama, entretanto, esse alinhamento entre sinalização e realidade metabólica é diferenciado. A célula tumoral opera sob um regime de glicólise-6-fosfato fermentativa acelerada, no qual a produção citosólica de ATP é elevada e sustentada, mantendo os pools energéticos globais em níveis compatíveis com processos anabólicos intensos. Nesse contexto, a ativação da AMPK ocorre predominantemente por vias não energéticas, especialmente pela elevação crônica do cálcio intracelular e ativação da CaMKKβ. Embora essa ativação preserve a capacidade catalítica da AMPK de fosforilar alvos, ela não representa um sinal estruturalmente coerente com um ambiente de escassez energética. A acetil-CoA carboxilase(ACC), portanto, recebe um sinal inibitório que não corresponde ao estado energetico real do metabolismo celular.
Em contraste, a sinalização da insulina ocorre em um contexto estruturalmente congruente com a função da acetil-CoA carboxilase(ACC). A insulina sinaliza abundância de carbono, disponibilidade energética e necessidade de armazenamento e biossíntese, exatamente o ambiente no qual a acetil-CoA carboxilase(ACC) tem um padão para operar. Dentro desse mesmo contexto a ativação do eixo PI3K–Akt pela insulina, que vai promover a ativação e o recrutamento local de fosfatases, principalmente a proteína fosfatase 2 (PP2A), que removem a fosforilação inibitória da acetil-CoA carboxilase(ACC) nos resíduos regulatórios críticos. Uma vez desfosforilada, a acetil-CoA carboxilase(ACC) reassume com uma conformação estrutural compatível com a polimerização, permitindo o alinhamento adequado de seus domínios catalíticos e a restauração do ciclo completo de transferência da biotina entre os sítios ativos. Além disso, a insulina favorece um ambiente citosólico rico em citrato e acetil-CoA, o que reforça a estabilidade estrutural da forma polimérica ativa da acetil-CoA carboxilase(ACC).
Nesse contexto o citrato no citosol, atua como modulador alostérico positivo, promovendo interações intermoleculares que estabilizam os filamentos enzimáticos, enquanto o acetil-CoA abundante garante ocupação eficiente do sítio catalítico. Embora esses fatores não sejam capazes de reverter a fosforilação imposta pela AMPK, eles são decisivos para consolidar a atividade máxima de atuação da acetil-CoA carboxilase(ACC) assim que a desfosforilação ocorre. Desse modo, a estrutura da acetil-CoA carboxilase(ACC) passa a refletir o ambiente metabólico que é verdadeiro da célula tumoral, e não um sinal energético errado, transmitido pela AMPK ativada por cálcio.
Esse comportamento estrutural explica por que, no câncer de mama, a insulina prevalece funcionalmente sobre a AMPK no controle da acetil-CoA carboxilase(ACC). Não se trata de uma simples competição entre uma quinase e uma fosfatase, mas de uma hierarquização baseada na coerência entre sinalização e um estado metabólico real. A acetil-CoA carboxilase(ACC) atua como um integrador estrutural que privilegia sinais compatíveis com alta disponibilidade energética e abundância de carbono, permitindo que a lipogênese de novo permaneça ativa mesmo na presença de uma AMPK funcional. Assim, a enzima não ignora a AMPK, mas vai responde preferencialmente ao sinal que melhor representa o ambiente verdadeiro no qual está inserida, consolidando a lipogênese como um dos eixos metabólico sustentado no câncer de mama.
Conclusão
No câncer de mama, a atividade da acetil-CoA carboxilase é determinada por uma hierarquia regulatória clara: a AMPK impõe um controle energético dominante por fosforilação, enquanto a insulina, por meio da ativação e redistribuição local da PP2A, remove esse freio e restaura a lipogênese. A abundância citosólica de acetil-CoA não ativa a acetil-CoA carboxilase(ACC) por si só, mas garante plasticidade metabólica, permitindo que haja uma rápida retomada do fluxo lipídico sempre que o sinal energético é localmente neutralizado. Esse arranjo representa uma adaptação bioquímica comprovada que sustenta a progressão tumoral no câncer de mama
Para compreender a ativação da lipogênese no câncer de mama, também é fundamental entender a estrutura da acetil-CoA carboxilase (ACC)
A acetil-CoA carboxilase (ACC) é uma enzima complexa e multifuncional, contendo três domínios principais (Biotina Carboxilase – BC, Carboxil Transferase – CT e Proteina Transportadora de Bioina – BCCP) dentro de um único polipeptídeo em eucariontes, trabalhando em homodímeros para catalisar a carboxilação da Acetil-CoA em Malonil-CoA, etapa chave na síntese de ácidos graxos, utilizando a biotina como cofator para transferir o CO2 entre os sítios ativos.
Estruturalmente a Acetil-CoA Carboxilase (ACC), tem 3 domínios catalítico-funcionais principais.
- BC (Biotin Carboxylase)
– catalisa a carboxilação da biotina
– contém o sítio de ligação ao ATP e ao bicarbonato
- BCCP (Biotin Carboxyl Carrier Protein)
– contém a lisina biotinilada
– funciona como o braço móvel que transporta a biotina - CT (Carboxyl Transferase)
– transfere o grupo carboxila da biotina para a acetil-CoA
Esses três são os domínios estruturais e catalíticos canônicos da acetil-CoA carboxilase (ACC).
Mecanismo Simplificado
- Carboxilação da Biotina: O domínio BC adiciona um CO2 (do bicarbonato) à biotina ligada ao domínio BB, formando carboxibiotina.
- Transferência: A biotina carboxilada gira (transloca) para o domínio CT.
- Formação do Malonil-CoA: No domínio CT, o CO2 é transferido para a Acetil-CoA, formando Malonil-CoA (o primeiro passo da síntese de ácidos graxos).
Dimerização e Regulação
- Para que a biotina da molécula de ACC alcance a Acetil-CoA na mesma molécula, as unidades de ACC formam homodímeros (duas moléculas de ACC juntas), permitindo a transferência do CO2 entre elas.
- A atividade da acetil-CoA carboxilase (ACC) é finamente regulada por fosforilação (por AMPK, por exemplo), que pode inibir a enzima, e por outros moduladores alostéricos.
Conclusão
A acetil-CoA carboxilase (ACC) é uma máquina molecular que coordena a adição de CO2 à Acetil-CoA, que essencial para a biossíntese de lipídios, com uma estrutura que facilita essa reação em cascata através de seus domínios funcionais e da formação de dímeros.
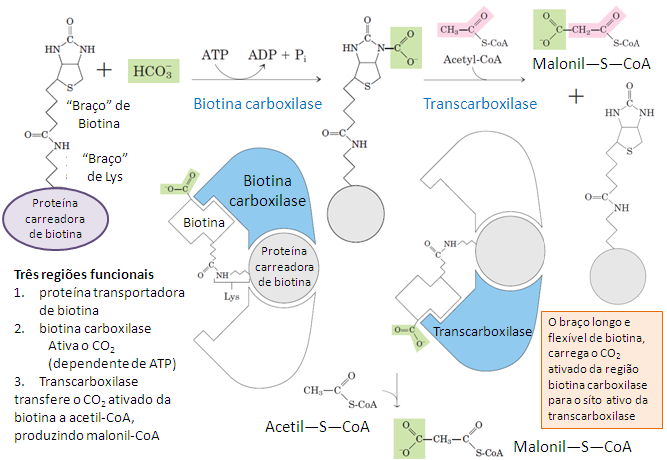
Ligação covalente da biotina (via lisina) na acetil-CoA carboxilase (ACC) no câncer de mama
A ligação covalente da biotina à lisina conservada do domínio BB (BCCP) não é regulada dinamicamente pela sinalização tumoral, pois essa ligação é catalisada pela holocarboxilase sintetase (HLCS), onde ocorre co-tradução ou logo após a tradução da acetil-CoA carboxilase(ACC) e uma vez biotinilada, a ACC permanece biotinilada por longos períodos
Conclusão importante que se deve levar em consideração nesse contexto.
No câncer de mama, o principal fator não é o aumento da ligação covalente da biotina, pois isso não é o passo regulatório da atividade enzimática. Mesmo que haja mais biotina disponível a aumento de HLCS, isso não explica o aumento funcional rápido da acetil-CoA carboxilase(ACC) tumoral.
O verdadeiro ponto regulatório do movimento da biotina (domínio BB/BCCP) é o que realmente aumenta a atividade da acetil-CoA carboxilase(ACC) no câncer de mama é a facilitação do movimento de translocação da biotina entre os sítios catalíticos (BC ↔ CT), mediada por mudanças conformacionais da enzima e esse movimento depende do estado estrutural da ACC, e não da presença da biotina em si.
Entre os principais fatores que favorecem o câncer de mama, destaca-se o aumento do citrato citosólico.
Pois este é o fator dominante na ativação da liogenese no câncer de mama, onde nesse contexto há um alto fluxo glicolítico + glutaminólise, com um aumento de citrato exportado da mitocôndria para o citosol onde o citrato liga-se diretamente à ACC1
Esse efeito estrutural do citrato induz a polimerização da acetil-CoA carboxilase (ACC) que estabiliza a forma ativa e alongada que aumenta a eficiência do “biotin swing” do domínio BB, isso aproxima fisicamente os domínios BC e CT onde o citrato não aumenta a ligação da biotina, mas aumenta drasticamente a capacidade funcional da biotina que já está ligada.
Outros Fatores secundários que reforçam esse efeito é a Insulina / PI3K–Akt, que ativa fosfatases (ex.: PP2A) removendo a fosforilações inibitórias da ACC isso favorece a conformação permissiva ao movimento do BB. A alta razão acetil-CoA/malonil-CoA não regula a biotinilação mais favorece o fluxo catalítico após o swing da biotina
A Inibição funcional da AMPK pela fosforilação da acetil-CoA carboxilase (ACC), bloqueia a mobilidade conformacional, mais no câncer, AMPK é frequentemente funcionalmente suprimida e sobreposta por sinais anabólicos
Conclusão
No câncer de mama, o fator que mais aumenta a atividade da acetil-CoA carboxilase (ACC) não é a ligação covalente da biotina à lisina, pois ela já é constitutiva.
Um dos principais fatores na ativação da acetil-CoA carboxilase (ACC), é o aumento do citrato citosólico, que promove polimerização e rearranjo conformacional da ACC, facilitando o movimento do domínio BB/BCCP e, portanto, a atividade enzimática.
Função diferenciada da BCCP na Acetil-CoA Carboxilase (ACC)
No contexto do câncer de mama, entre os três componentes funcionais da acetil-CoA carboxilase, destaca-se aquele cuja função é mais profundamente modificada.
1 – Biotin Carboxylase (BC)
2 – Carboxyl Transferase (CT)
3 – Biotin Carboxyl Carrier Protein (BCCP), é o domínio que apresenta a função mais diferenciada e explorada pela célula tumoral é o BCCP.
Em condições fisiológicas, os três domínios operam de forma coordenada e relativamente estável, mas na célula tumoral mamária a reprogramação metabólica não altera de modo significativo a química básica da reação catalisada por BC (carboxilação da biotina dependente de ATP) nem por CT (transferência do grupo carboxila da biotina para o acetil-CoA); essas etapas permanecem bioquimicamente conservadas. O que muda de forma mais marcante é a dinâmica estrutural da enzima, e é nesse ponto que o BCCP assume papel central.
O BCCP funciona como um braço móvel que transporta a biotina covalentemente ligada entre os sítios ativos de BC e CT, e sua eficiência depende diretamente da conformação global da ACC. No câncer de mama, o aumento do citrato citosólico, a ativação de vias anabólicas (como insulina/PI3K–Akt) e a atenuação funcional da AMPK favorecem a polimerização e a conformação ativa da ACC, ampliando a mobilidade e o alcance funcional do domínio BCCP. Dessa forma, a biotina já ligada passa a “trabalhar melhor”, com maior frequência e eficiência de translocação, acelerando o fluxo catalítico total da enzima sem necessidade de modificar a reação química em si.
Assim, embora BC e CT sejam indispensáveis e cataliticamente ativos, suas funções não são qualitativamente redefinidas no câncer de mama; elas apenas acompanham o aumento de fluxo imposto pela reorganização estrutural da enzima. O BCCP, por outro lado, torna-se o principal ponto de diferenciação funcional, pois é ele que traduz o ambiente metabólico tumoral em maior eficiência catalítica, conectando diretamente a reprogramação metabólica à ativação sustentada da lipogênese tumoral.
Quimicamente e estruturalmente, o BCCP é menos influenciado pela AMPK no câncer de mama porque ele não é um domínio catalítico nem um alvo direto de fosforilação regulatória, e sua função depende muito mais de propriedades mecânicas e geométricas da proteína do que de modificações químicas locais.
Em primeiro lugar, a AMPK reconhece motivos específicos de fosforilação em resíduos de serina/treonina inseridos em contextos estruturais definidos. Na ACC, esses motivos estão concentrados principalmente fora do BCCP, em regiões regulatórias que controlam a oligomerização e o estado conformacional global da enzima. O BCCP, por sua função essencial de carregar a biotina, é quimicamente conservado e estruturalmente compacto, com uma lisina biotinilada enterrada em um dobramento estável, pouco acessível a quinases. Fosforilar diretamente o BCCP comprometeria o encaixe da biotina e a viabilidade catalítica da enzima, razão pela qual a evolução “protegeu” quimicamente esse domínio de regulação por AMPK.
Em segundo lugar, a função do BCCP é transitória e cinética, não química: ele atua como um braço flexível que oscila entre os sítios BC e CT. A AMPK não altera a química da ligação biotina–lisina nem a energia do “swing” do BCCP; ela atua reduzindo a atividade da ACC ao desfavorecer a polimerização e a conformação alongada da enzima. No câncer de mama, o aumento do citrato citosólico e a sinalização anabólica estabilizam essa conformação ativa, neutralizando o impacto conformacional da fosforilação induzida pela AMPK. Assim, mesmo quando a ACC é parcialmente fosforilada, o ambiente químico favorece a mobilidade funcional do BCCP.
Além disso, do ponto de vista químico-energético, a atividade do BCCP não é um passo limitante dependente de ATP ou de potencial redox — ao contrário do domínio BC, que consome ATP e é altamente sensível ao estado energético celular, o principal sensor da AMPK. Como o BCCP apenas transporta um grupo químico já ativado, ele escapa do controle direto da AMPK, cuja lógica regulatória é poupar energia bloqueando reações energeticamente custosas.
Por fim, no câncer de mama, a AMPK tende a atuar de forma local e incompleta, enquanto o citrato, a insulina e o alto fluxo de substratos impõem um controle estrutural contínuo. Quimicamente, isso significa que a modulação conformacional global supera a modificação covalente pontual, tornando o BCCP funcionalmente resiliente à inibição mediada pela AMPK.
Em síntese, o BCCP é menos influenciado pela AMPK porque não contém alvos químicos regulatórios diretos, sua função é mecânica e conservada, não energeticamente custosa, e porque, no câncer de mama, o controle estrutural imposto pelo citrato e por sinais anabólicos domina sobre a fosforilação inibitória global da ACC.
Quimicamente
(quimicamente há uma hierarquia de níveis regulatórios).
O BCCP é menos influenciado pela AMPK ao nível químico direto. Porque a AMPK não fosforila o BCCP, não altera a ligação biotina–lisina e não modifica a energia intrínseca do movimento do braço biotinilado. Nesse sentido estrito, o BCCP é quimicamente pouco sensível à AMPK.
Mais o BCCP como o domínio funcionalmente é mais alterado no câncer de mama, em nível funcional-integrativo, não químico. O câncer não muda o BCCP em si, mas muda o ambiente estrutural da ACC (citrato elevado, polimerização, sinalização anabólica), e quem mais se beneficia dessa reorganização é exatamente o BCCP, porque a sua função depende da geometria e da dinâmica global da enzima.
- BC continua carboxilando a biotina usando ATP (passo químico conservado e energético).
- CT continua transferindo o grupo carboxila para o acetil-CoA (passo químico conservado).
- BCCP, porém, deixa de ser apenas um “transportador passivo” e passa a ser o principal ponto de amplificação funcional, porque o câncer de mama reorganiza a estrutura da ACC (via citrato, insulina e supressão funcional da AMPK), aumentando drasticamente a eficiência, frequência e alcance do movimento da biotina.
Em termos conceituais
A AMPK regula a ACC freando a máquina, mais os citrato e os sinais anabólicos reorganizam a máquina. Neste assundo é o BCCP a peça que mais muda de desempenho quando a máquina é reorganizada, mesmo sem ser quimicamente modificada.
Assim, no câncer de mama, o domínio menos regulado quimicamente pela AMPK é exatamente aquele que mais é expressa funcionalmente na reprogramação metabólica, o que explica por que o BCCP aparece simultaneamente como pouco sensível à AMPK e central para o aumento de atividade da ACC.
Conclusão
A AMPK regula a ACC freando a máquina, mais os citrato e os sinais anabólicos reorganizam a máquina. Neste assundo é o BCCP a peça que mais muda de desempenho quando a máquina é reorganizada, mesmo sem ser quimicamente modificada.
Assim, no câncer de mama, o domínio menos regulado quimicamente pela AMPK é exatamente aquele que mais é expressa funcionalmente na reprogramação metabólica, o que explica por que o BCCP aparece simultaneamente como pouco sensível à AMPK e central para o aumento de atividade da ACC.
Deus o pai diz (Provérbios 20:18)
Os conselhos são importantes para quem quiser fazer planos, e quem sai à guerra precisa de orientação.
Provérbios 20:18
Referência:
Alteração da fosforilação no câncer por meio de modificadores da PP2A
https://link.springer.com/article/10.1186/s12935-023-03193-1
Proteína fosfatase 2A: um alvo para terapia anticancerígena
https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(12)70558-2/abstract
Identificação de complexos PP2A específicos envolvidos na transformação de células humanas.
https://www.sciencedirect.com/science/article/pii/S1535610804000261
Proteína fosfatase 2
https://en.wikipedia.org/wiki/Protein_phosphatase_2
A desregulação do inibidor da fosfase de proteina 2A, SET está associada á progreção maligna do câncer de mama
https://www.nature.com/articles/s41598-021-93620-y
Acetil-CoA carboxilase
https://pt.wikipedia.org/wiki/Acetil-CoA_carboxilase
Acetil-CoA Carboxilases e Doenças
https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2022.836058/full
Metabolismo de Lipídios e Câncer
https://www.mdpi.com/2075-1729/12/6/784
contínua no próximo post
A cura será apresentada no final de 250 posts